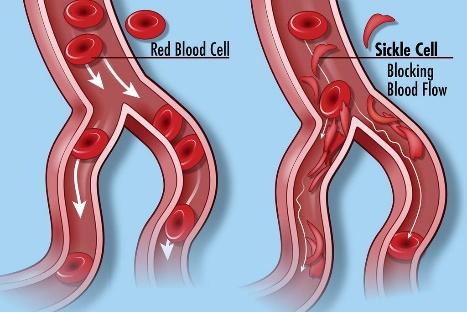
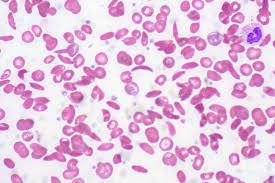
Living with sickle cell disease (SCD) presents numerous challenges that affect individuals not only physically but also psychologically. The unpredictable nature of pain crises, the constant requirement for symptom management, and the emotional toll of chronic illness collectively contribute to heightened levels of stress, anxiety, and depression in those with SCD. To cope with these mental health challenges, individuals with SCD may turn to alcohol as a form of self-medication, seeking relief from persistent physical discomfort, frequent hospitalizations, limitations in daily activities, and the uncertainty surrounding the future associated with SCD. The potential for complications and the profound impact on overall quality of life further intensify psychological distress. Consequently, some individuals grapple with the emotional toll of SCD, and the appeal of alcohol arises as a perceived means to navigate and alleviate the burdens imposed by the condition (Bruton, 2016).
The American Psychological Association (APA) underscores the significance of moderate alcohol use, defining it as no more than two drinks daily for men and one for women and older individuals. While generally considered harmless for most adults, this seemingly moderate approach signals the initiation of a continuum stretching from alcohol abuse to alcohol dependence. Such a progression can have a profound impact, especially for individuals confronting the distinctive challenges of sickle cell disease. Navigating this challenge, individuals contending with alcoholism or alcohol dependence experience growing difficulty in controlling their consumption. They grapple with the challenge of halting the process once initiated, and this dependency leads to increased tolerance, demanding higher quantities for the same desired effect (Alao et al., 2003; Levenson et al., 2007).
Approximately 30% of people with sickle cell disease grapple with symptoms of depression (Bruton, 2016; Levenson et al., 2008). Alcohol is a depressant that can worsen depression in the long run. It increases depression symptoms, causes mood swings, disrupts sleep, reduces the quality of sleep, interferes with many medications, especially antidepressants, increases risk-taking behaviors, and can diminish physical health. Initially, it may lead to feelings of relaxation and temporary increases in confidence, but these are swiftly followed by negative emotions such as anger, depression, and anxiety due to their impact on the inhibition control center.

This disruption hampers cognitive processes, impeding the genuine interpretation of feelings and the evaluation of potential consequences. This impact is more pronounced in individuals with sickle cell disease (SCD). Additionally, sustained alcohol use in the Sickle Cell population may have long-term effects, depleting neurotransmitter levels crucial for combating anxiety and depression. This depletion can foster a heightened desire for alcohol consumption, initiating a cycle of dependence that can amplify the vulnerability to impulsive behaviors, often leading to suicide and death (Valenzuela, M.D., Ph.D., Year; American Addiction Centers, 2024) Alcohol’s capacity to diminish inhibitions and enhance impulsivity may lead individuals to engage in actions such as self-harm or suicide. ((Larkin et al., 2017; Pennel et al., 2015; Haw, C. et al., 2005; Alcohol and Mental Health, n.d).
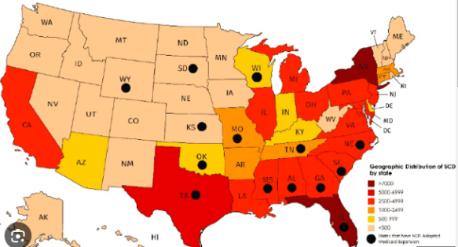
The intricate relationship between sickle cell disease (SCD) and mental health, particularly alcohol abuse, underscores the interconnected nature of physical and mental well-being within the SCD population. According to Levenson et al. (2008), 31.4% of adults with SCD are identified as alcohol abusers, indicating a relatively high prevalence of alcohol misuse compared to the national average of 48.7% (SAMHSA, 2023). Simultaneously, SCD is associated with a notable prevalence of depression, ranging from 18% to as high as 44%, surpassing rates observed in the general population (Levenson, 2008). Emphasizing this connection further, among those with anxiety (7%) within the SCD population, a striking 83% also suffer from depression, contributing to an overall depression prevalence of 28%.
While individuals with sickle cell disease confront the dual challenge of managing both physical pain and emotional distress, resorting to alcohol as a coping mechanism is problematic. The short-lived relief and subsequent rebound effect can contribute to a cycle of dependency, worsening pre-existing mental health conditions such as depression (American Psychological Association, 2012), which are already heightened due to the chronic nature of the disease. Additionally, alcohol can induce further challenges, including memory loss and increased anxiety, ultimately complicating the emotional well-being of these individuals.
As previously mentioned, alcohol negatively affects sleep in a population that already experiences disruptions due to pain and discomfort (Stein, M.D. & Friedmann, P.D., 2008; Vitiello, M. V., 2006). Furthermore, a significant 44% of this population suffers from sleep-disordered breathing (Sharma et al., 2015). Although alcohol may initially induce sedation, its interference with sleep patterns can exacerbate fatigue, irritability, and difficulties in managing both the physical and emotional aspects of the disease. This impact on sleep can also extend to cognitive function, impairing memory, and concentration. Those already facing cognitive challenges due to sickle cell disease (SCD) may experience further disruption from alcohol use. Additionally, with 57% of this population reporting complaints of insomnia, their struggles are intensified. This compounded impact can potentially lead to feelings of frustration and helplessness (Mann-Jiles et al., 2015; Sharma et al., 2015).

Patients with sickle cell disease (SCD) face a heightened risk of 30-day readmissions, with the median hospital stay typically being 5 days on average (Bruton, 2016; Silverstein et al., 2020). This heightened readmission risk exposes SCD patients to a series of acute care interventions during hospital stays, including repeated blood draws, x-rays, blood transfusions, and inadequately treated pain. These unpleasant experiences, coupled with inconsistent and unfriendly interactions within the healthcare system, contribute to a cycle of heightened stress and depression (Pecker & Darbari, 2019). Furthermore, the financial ramifications of recurrent hospitalizations add to the overall burden on individuals grappling with sickle cell disease, further worsening the situation. In fact, among the SCD outpatient population, a mental health diagnosis is linked to a higher median cost of care. This suggests that SCD patients with documented mental health conditions typically face increased expenses for outpatient care (Bruton, 2016).
The social consequences of alcohol use not only heighten the psychological burden for individuals with Sickle Cell Disease (SCD) but also significantly impact their family members. This is underscored by the understanding that alcohol problems don’t just hurt the drinker (APA, n.d). In individuals with SCD, alcohol-related behaviors can strain familial relationships, lead to social isolation, and impair decision-making abilities. This not only deepens the emotional toll on the individuals themselves but also on their caregivers, who may feel overwhelmed by witnessing the challenges posed by alcohol use. As a result, the mental health struggles of both parties are exacerbated, creating a challenging cycle that is difficult to break.

Addressing the psychological implications of alcohol use in patients with sickle cell disease is a critical component of comprehensive care for those living with this chronic condition. Recognizing the motivations behind alcohol consumption and understanding its impact on both physical and mental health are essential for implementing effective intervention strategies. These strategies are fundamental in laying the groundwork that will help in improving the overall well-being of individuals affected by SCD. By acknowledging and addressing this complex relationship, healthcare professionals and other stakeholders can contribute to a more holistic and patient-centered approach to managing sickle cell disease.